X-ray powder diffraction (XRPD) is one of the most powerful methods for the study of crystalline and partially crystalline solid state materials. Each crystalline form has a unique powder diffraction fingerprint that can be used to identify the presence of that form within a sample. We have extensive experience in the interpretation of powder diffraction patterns and have developed unique methodologies for fully automated recognition of crystalline forms.
As a crystalline form becomes disordered, it may pass through microcrystalline and nanocrystalline forms before reaching a final amorphous or random form. Like the crystalline form, each of these disordered forms has a unique diffraction fingerprint that we can extract with our proprietary pattern recognition software, enabling each form’s identification within a sample. The relative amounts of each of these unique diffraction patterns can be used to quantify sample composition. We have a range of controlled humidity and temperature chambers where the thermodynamics of these various crystalline and disordered forms can be studied in situ and in real time.
We offer various sample presentation methods allowing samples to be studied in solid, powder, suspension, cream or melt form. Our expert scientists have developed approaches to overcome common sample limitations such as inhomogeneity, preferred orientation and particle statistics, and extremely small amounts of material can be analyzed using these techniques.
Indexing
X-ray powder diffraction pattern indexing is the first step in the solution of a crystal structure from powder data but can also be used to determine if a given pattern represents a pure solid phase. The latter information is often sufficient to ensure product homogeneity if a crystal structure is unavailable. Our patented algorithm is an essential tool in the determination and confirmation of crystalline phase purity.
Single Crystal X-Ray
An unequivocal method of solid form identification, and one preferred by the FDA when possible, is single crystal X-ray structure determination. An attempt should be made to obtain a single-crystal structure on all suitably crystalline new drug candidates. Our scientists employ the most modern methods, including a state-of-the-art Rigaku SuperNova diffractometer equipped with a micro-focus X-ray source and HPAD detector. This allows the determination of structures using smaller crystals than are required for conventional equipment.
Calculation of Powder Patterns
Calculation of an X-ray powder diffraction pattern from single crystal data provides important information about the homogeneity of a crystalline solid, as well as an unequivocal pattern for use as a standard. We use multiple software methods to calculate powder patterns from crystal coordinate files.
Once the crystal structure of a material has been determined, we can calculate an XRPD pattern. The calculated powder pattern provides information about homogeneity of the crystalline phase and serves as an unequivocal pattern for use as a standard for polymorph screening or quantitative analysis of multiphase samples. We can build parameters such as crystallite size, morphology and preferred orientation into the pattern calculation, providing insight into the microstructure of single-phase samples. Microstructural parameters often play a significant role in the physical and chemical properties of a sample.
One of the more powerful features of pattern calculation is the ability to study disordering processes to identify relationships between crystalline phases and microcrystalline/amorphous phases. The subsequent calculated amorphous patterns can be used for precise crystallinity analysis if pure amorphous forms of the material are not available for measurement. We offer a wide range of tools for the calculation of powder patterns starting from crystal coordinate files, D and I files or reference patterns.
Rietveld Analysis
The Rietveld method is a computational treatment of diffraction data that separates overlapping data from a typical XRPD pattern and allows accurate determination of the structure of a compound. We use this method to address various problems including quantitation using XRPD patterns with high degrees of overlap.
Crystal Structure Determination From X-Ray Powder Diffraction
On occasion, suitable single crystals cannot be grown for a given molecule or particular solid form thereof. If a good quality powder sample is available, we can often determine the crystal structure using computational methods.
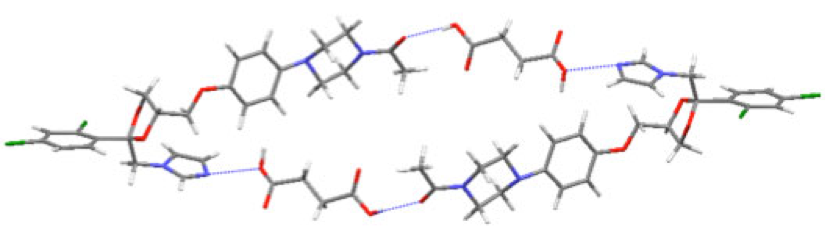
Hydrogen bonding motif in the ketoconazole: succinic acid cocrystal as determined through X-ray powder diffraction.
We have a long track record of solving structures from powder data. Although we have experience with synchrotron data, in most cases good results can be obtained using our in-house, high-resolution XRPD data.
We perform a structure solution using a computational method called simulated annealing, in which the molecular structure is used as the input model and the conformation, orientation and position of the molecule is varied until a plausible solution is found. Larger, flexible molecules and multicomponent crystals require more computational time. We have a parallel implementation for structure solution. The more complicated ketoconazole cocrystals showcased in the application note were solved in overnight calculations.
The major caveat, in comparison to structure determination from single crystals, is that the absolute configuration cannot be determined from powder data due to the overlap of the Friedel pair intensities needed for that analysis. We recommend performing additional experimental testing to confirm the molecular structure and the composition in case of solvated crystals using vibrational spectroscopy.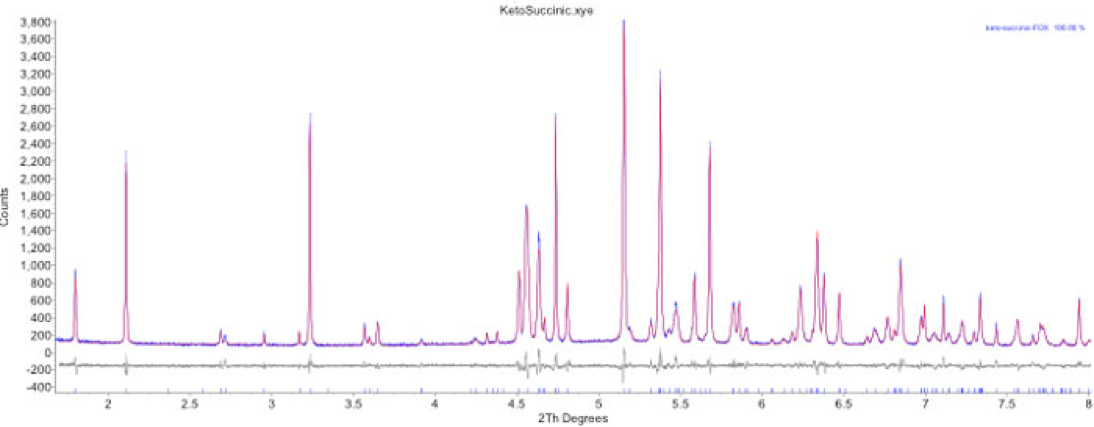
Final Rietveld refinement result for the ketoconazole: succinic acid cocrystal.
Contact our solid state services team at SSCI by clicking below